
news
New Breakthrough Changes the Shape of Treatment for Undruggable Diseases
For some time, scientists have been working on the major challenge of developing new therapies against many human diseases. Many of these diseases are caused by the aberrant action of certain proteins in our cells that are considered “undruggable”, or difficult to therapeutically target using classical drug discovery methods. A major class of “undruggable” proteins are those that are aberrantly degraded and destroyed in the cell leading to many human diseases, including cancer, neurodegenerative diseases, metabolic diseases, and hundreds of genetic disorders.
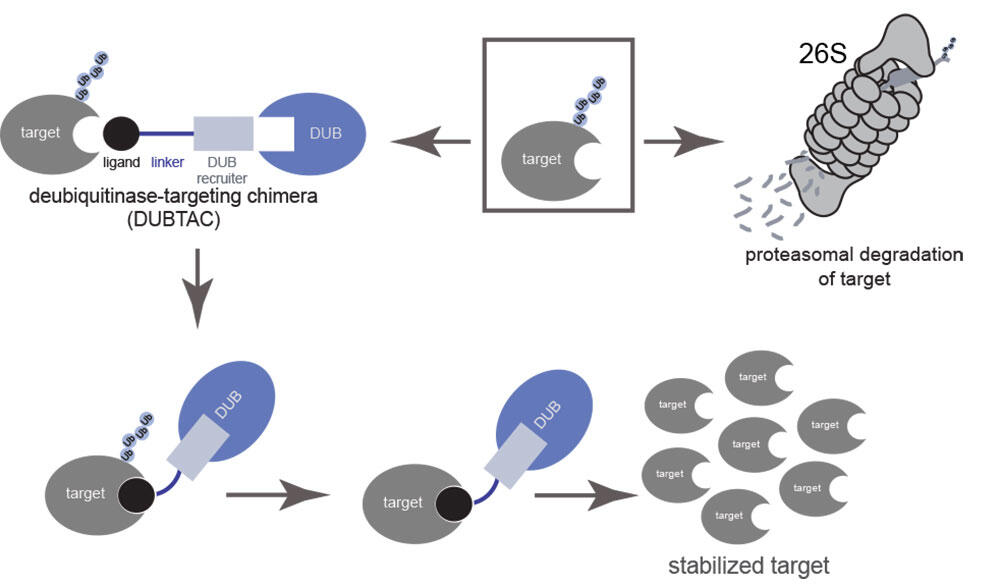
These proteins are frequently destroyed through tagging with chains of a protein known as ubiquitin, which signals the cell to destroy the proteins through the “cellular trash can” called the proteasome.
While these proteins would benefit therapeutically by stopping their destruction and stabilizing and increasing the levels of the actively destroyed proteins, targeted protein stabilization of an actively degraded protein with a small-molecule drug has thus far remained impossible or “undruggable”.
Now, a team of researchers from the lab of Daniel Nomura, Professor of Chemical Biology and Director of the Novartis-Berkeley Center for Proteomics and Chemistry Technologies at UC Berkeley and IGI researcher, in collaboration with researchers from the Novartis Institutes for BioMedical Research have reported the discovery of a new therapeutic paradigm called Deubiquitinase Targeting Chimeras (DUBTACs) to address the challenge of targeted protein stabilization.

DUBTACs are dumbbell shaped molecules which consist of a chemical compound that binds to the disease-causing protein linked via a linker to another chemical recruiter of an enzyme called deubiquitinase or DUB. The DUB removes the ubiquitin chains from actively degraded proteins, thus preventing their destruction and stabilizing their protein levels.
In new research published in Nature Chemical Biology, the researchers report on how DUBTACs stabilize the mutant form of the CFTR chloride channel protein that is otherwise degraded to cause the debilitating disease cystic fibrosis. Cystic fibrosis was chosen as the initial test case for DUBTACs as this disease is caused by mutations in the CFTR channel which cause this protein to become unstable and actively degraded, leading to the cystic fibrosis pathology. The researchers postulated that a DUBTAC against mutant CFTR could stabilize and increase the levels of this mutant protein to restore CFTR function in cystic fibrosis patients.
The research has demonstrated that the CFTR DUBTAC restores chloride channel function in primary human bronchial epithelial cells from cystic fibrosis patients compared to currently approved therapies. The researchers also showed that a DUBTAC could be applied to cancers as well. They developed a DUBTAC to stabilize a tumor suppressor protein WEE1 kinase. WEE1 kinase is actively degraded in many tumors to promote cancer cell proliferation. Stabilizing the levels of WEE1 kinase in cancer cells could stop tumor growth.
Prof. Nomura states, “We believe that this new DUBTAC therapeutic platform can be used to develop a new class of medicines against many human diseases, including cancer, neurodegenerative diseases, and many genetic disorders by enabling access to these previously “undruggable” proteins that were actively degraded to drive these diseases.”
The lead authors for this research are UC Berkeley chemical biology graduate students Nathaniel Henning and Lydia Boike. Additional co-authors include Jessica Spradlin, Carl Ward, Gang Liu, Erika Zhang, Bridget Belcher, Scott Brittain, Matthew Hesse, Dustin Dovala, Lynn McGregor, Rachel Valdez Misiolek, Lindsey Plasschaert, David Rowlands, Feng Wang, Andreas Frank, Daniel Fuller, Abigail Estes, Katelyn Randal, Anoohya Panidapi, Jeffrey McKenna, John Tallarico, and Markus Schirle from UC Berkeley and the Novartis Institutes for BioMedical Research.
This story was originally published by the UC Berkeley College of Chemistry.
Learn more:
Deubiquitinase-targeting chimeras for targeted protein stabilization.
Media contacts:
Andy Murdock, andymurdock@berkeley.edu
Daniel Nomura, dnomura@berkeley.edu


