Introduction How do scientists use CRISPR tools in the lab?
Scientists have taken natural CRISPR proteins and reimagined them as tools for manipulating the genome in other organisms. This CRISPRpedia chapter describes a variety of CRISPR-based tools, how they work, and how they can be used for research.
Cas9: From nature to the lab
The bacterial Cas9 protein has been co-opted as a genome-editing tool for use in any organism
- Vocabulary
- CRISPR RNA/crRNA, dual guide RNA/dgRNA, single guide RNA/sgRNA, nucleus, eukaryotes, prokaryotes, nuclear localization signal/NLS, histones
Two different uses for programmable DNA cutting
As discussed in the previous chapter, there are many natural CRISPR systems that protect bacteria from viruses. But one CRISPR system in particular has opened the door to a new era in biological research. This is CRISPR-Cas9, which has been repurposed as a tool for altering targeted DNA sequences in a process called genome editing. Before we dive into the details of how genome editing works, we'll review how Cas9 works in nature and what's different about using it as a genome-editing tool. The core components involved in both CRISPR immunity and genome editing are:
- The Cas9 protein — molecular scissors that cut DNA
- A guide RNA — a small, customizable molecule that directs Cas9 to cut a specific DNA sequence
Both bacterial immunity and genome editing depend on Cas9's ability to find and cut specific DNA targets based on the sequence of its guide RNA. Let's look through some important differences between these two contexts: delivery, guide RNA composition, Cas protein variants, and prokaryotic vs. eukaryotic cells.
Delivery
How does the Cas9 get into a cell? In nature, bacterial DNA encodes the CRISPR-Cas9 system. But Cas9 is not naturally encoded in complex organisms like plants or humans, nor in all microbes that we might like to study, so researchers must add or "deliver" the system to these organisms for genome editing. To do so, they must bypass cellular barriers. Researchers bypass these barriers in a variety of ways, depending on which type of organism they're working with and exactly what they're trying to do.
Some common ways to add CRISPR tools to cells in the lab include transforming plasmids (for bacteria), electroporation, chemical stimulation, or viral delivery (for human cells), Agrobacterium transformation (for plants), and microinjecting eggs or embryos (for animal models like mice or zebrafish).
Guide RNA composition
In nature, Cas9 is guided by two separate RNA molecules, sometimes called a dual-guide RNA (dgRNA):
- A CRISPR RNA (crRNA) that dictates what site Cas9 will cut
- A trans-activating CRISPR RNA (tracrRNA) that attaches to the crRNA and acts as a handle for Cas9
Researchers found a way to simplify this. They combined the crRNA and the tracrRNA to form one, continuous molecule known as a single-guide RNA (sgRNA). As a result, genome editors only need to generate one RNA to edit a particular DNA sequence.
Cas protein variants
Bacterial cells will use whichever CRISPR system they have in their fight against phages, but researchers are not limited to using just one. The basic CRISPR-Cas9 system can do a lot, but it has limitations. Recognizing this, researchers have spent a lot of time developing CRISPR tools: first, they've made more accurate versions for genome editing. They've developed other Cas enzymes for genome editing and engineered Cas proteins to cut targets with new PAM sequences. They’ve even developed CRISPR systems that do things besides cut DNA. You can learn more about these changes in subsequent sections.
Prokaryotic vs. eukaryotic cells
Cas9 structure-function
How structural changes in the Cas9 nuclease let it cut target DNA
- Vocabulary
- Structure, domain, recognition lobe/REC, nuclease lobe/NUC, HNH, RuvC, PAM-interacting domain/PID, target strand, non-target strand, cleaving, off-target effect
A tour of the Cas9 protein and how it works
CRISPR nucleases like Cas9 find and cut DNA targets in a multi-step process. Each protein has a specific structure — a shape or architecture — that lets it do its job. A guide RNA directs each CRISPR nuclease to target DNA or RNA sequences. Note that since RNA is written in the same genetic "language" as DNA, RNA and DNA can base-pair with each other, and a guide RNA can pair with complementary DNA or RNA.
The nuclease has various built-in quality control steps that ensure that it only cuts the correct DNA sequence. Here, we describe how this process works for Cas9. Many CRISPR enzymes follow similar principles, but each has its own quirks, as detailed in the previous chapter.
Cas9 is a single, two-lobed protein made of several dynamic regions called domains. You can think of protein domains like different parts of the body — they're all connected and work together, but each does different things. Cas9 is bi-lobed because its domains form two lobes: the recognition lobe (abbreviated REC) and the nuclease lobe (NUC). These are sort of like the "top" and "bottom" of Cas9. In this way, its shape roughly resembles a clam.
The REC lobe is important for carrying the guide RNA. The NUC lobe contains two separate nuclease domains called HNH and RuvC. You can think of them as the two blades of the Cas9 "molecular scissors." The HNH domain is quite large and mobile, while the RuvC domain is less dynamic.
Next, we’ll walk through how each part of Cas9 helps it recognize and cut DNA.
Identifying the right DNA target
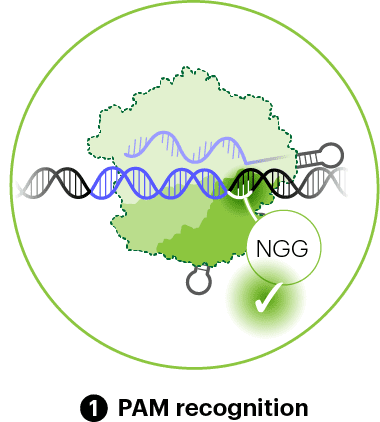 Cas9 carries a guide RNA, which is held mostly in its REC lobe. The Cas9–gRNA complex searches for matching DNA sequences. The first step in the search process is to look for a PAM, a short stretch of DNA that is required for Cas9 to bind targets and cut (explained in detail in the previous chapter). The PAM is identified by a region of the nuclease lobe called the PAM-interacting domain (PID). The widely-used Cas9 protein from the bacterium Streptococcus pyogenes recognizes the PAM sequence, “NGG.” Other CRISPR nucleases bind to unique PAMs because they have different amino acids in their PAM-interacting domains.
Cas9 carries a guide RNA, which is held mostly in its REC lobe. The Cas9–gRNA complex searches for matching DNA sequences. The first step in the search process is to look for a PAM, a short stretch of DNA that is required for Cas9 to bind targets and cut (explained in detail in the previous chapter). The PAM is identified by a region of the nuclease lobe called the PAM-interacting domain (PID). The widely-used Cas9 protein from the bacterium Streptococcus pyogenes recognizes the PAM sequence, “NGG.” Other CRISPR nucleases bind to unique PAMs because they have different amino acids in their PAM-interacting domains.
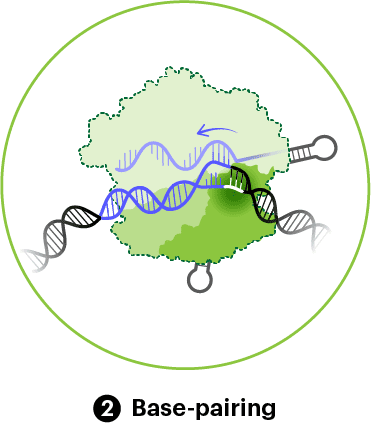 Once bound to a PAM, Cas9 unwinds the adjacent DNA double helix. If the guide RNA matches the target, it will zip up or base-pair with the complementary DNA strand (the target strand). Base-pairing always happens in one direction, starting from the bases right next to the PAM, known as the seed region, and zipping up all the way to end of the guide RNA. The base-paired gRNA and DNA target strand are cradled in the space between the REC and NUC lobes. Cas9 hangs onto the opposite or non-target strand in a groove along its surface, keeping the two DNA strands apart.
Once bound to a PAM, Cas9 unwinds the adjacent DNA double helix. If the guide RNA matches the target, it will zip up or base-pair with the complementary DNA strand (the target strand). Base-pairing always happens in one direction, starting from the bases right next to the PAM, known as the seed region, and zipping up all the way to end of the guide RNA. The base-paired gRNA and DNA target strand are cradled in the space between the REC and NUC lobes. Cas9 hangs onto the opposite or non-target strand in a groove along its surface, keeping the two DNA strands apart.
If there is no PAM or the adjacent DNA does not fully match the guide RNA, Cas9 will move on and search elsewhere.
The cutting process
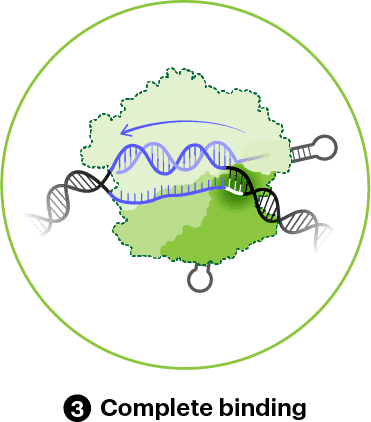 If Cas9 has found a PAM next to a region of DNA that matches its guide RNA, the process of cutting or cleaving the DNA will begin. Generally, Cas9 only cuts after all 20 nucleotides of the guide RNA base-pair with the target DNA. Cas9 occasionally ignores a few mismatches, leading to off-target cleavage. Read about efforts to improve Cas9 accuracy in “Improving CRISPR tools."
If Cas9 has found a PAM next to a region of DNA that matches its guide RNA, the process of cutting or cleaving the DNA will begin. Generally, Cas9 only cuts after all 20 nucleotides of the guide RNA base-pair with the target DNA. Cas9 occasionally ignores a few mismatches, leading to off-target cleavage. Read about efforts to improve Cas9 accuracy in “Improving CRISPR tools."
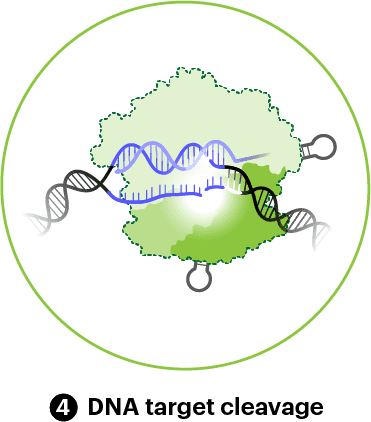 Once the target DNA is mostly or fully base-paired, the HNH domain swings into place and cuts the target DNA strand. Almost immediately after, the RuvC domain positions itself to cut the non-target DNA strand. The two cuts occur three nucleotides away from the PAM. They leave behind blunt DNA ends. However, the RuvC domain will often “chew” back on the non-target strand, leaving small overhangs.
Once the target DNA is mostly or fully base-paired, the HNH domain swings into place and cuts the target DNA strand. Almost immediately after, the RuvC domain positions itself to cut the non-target DNA strand. The two cuts occur three nucleotides away from the PAM. They leave behind blunt DNA ends. However, the RuvC domain will often “chew” back on the non-target strand, leaving small overhangs.
After cleaving the DNA, Cas9's job is done. In natural phage defense, other enzymes are thought to fully break down the cleaved phage DNA. During genome editing, cellular repair proteins mend the gap.
To review an animated version of this mechanism, check out HHMI's interactive Cas9 module. To get a closer look at the structure of Cas9, try our free augmented reality app, CRISPR-3D.
CRISPR genome editing
How making a cut in DNA causes a change in the sequence at the cut site
- Vocabulary
- Genome editing, zinc-finger nucleases/ZFNs, transcription activator-like effector nucleases/TALENs, double-strand break/DSB, non-homologous ending joining/NHEJ, indels, knock out, homology-directed repair/HDR, donor DNA/template DNA, on-target effects
How do scientists rewrite DNA?
Genome editing is the process of changing a specific DNA sequence within a living cell. This could be used for all sorts of purposes, described throughout CRISPRpedia.
There are multiple genome-editing tools available. For example, zinc-finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) are effective genome-editing tools that came before CRISPR. Researchers must re-engineer these tools each time they want to edit a new sequence. In contrast, CRISPR genome-editing technologies work in a "plug and play" way, making them much more convenient.
How does genome editing work? There are two key processes: (1) making a targeted cut in genomic DNA and (2) the cell repairing that DNA break in a way that changes the sequence.
Making a targeted DNA break
 The first step in editing the genome with CRISPR is to program Cas9 (or another Cas enzyme) with a guide RNA. Researchers design the guide to be complementary to a particular target sequence in the genome. They add Cas9 and their custom gRNA to the cells they want to edit. Cas9 will search the whole genome, eventually finding its target site and making a cut in both strands of DNA, also called a double-strand break (DSB).
The first step in editing the genome with CRISPR is to program Cas9 (or another Cas enzyme) with a guide RNA. Researchers design the guide to be complementary to a particular target sequence in the genome. They add Cas9 and their custom gRNA to the cells they want to edit. Cas9 will search the whole genome, eventually finding its target site and making a cut in both strands of DNA, also called a double-strand break (DSB).
CRISPR-Cas9, ZFNs, and TALENs are all genome-editing technologies that cut DNA wherever a researcher wants. This is why they're often described as "molecular scissors." But how can a DNA break end up changing the DNA sequence? Let’s walk through the rest of the process.
Two common ways to repair DNA breaks
Making a double-strand break in DNA is just the first half of genome editing. It’s up to the cell to do the rest. Damage to DNA can kill a cell, so cells have a variety of different repair systems in place to fix any issues. Making a DSB triggers the cell to send in a team of DNA repair proteins to mend the break.
The most common way for repair proteins to close a DNA break is to stitch the two broken ends back together. This often occurs through a process called non-homologous ending joining (NHEJ). The process of NHEJ can be sloppy, resulting in small DNA deletions or insertions at the repair site, abbreviated indels. These can inactivate or knock out genes.
Imagine deleting a word from the middle of a sentence or adding in a few random letters. The sentence probably won’t make sense anymore. Similarly, a gene containing an indel will usually stop working. After breaking a gene with NHEJ, scientists can observe the effects of the knockout and thereby learn about what the gene does. It can also be useful to stop genes from working when they have negative effects on an organism, like a gene variant that causes human disease or makes a plant susceptible to viral infection.
One of the less common repair mechanisms is for cells to fix DNA breaks by adding in new DNA sequences. This occurs through a process called homology-directed repair (HDR). With this process, researchers can specify exactly what DNA sequence gets inserted. They usually do so by delivering a segment of template or donor DNA. For effective repair, template DNA must contain sequences matching the broken DNA ends, called homologous sequences. Inserting or replacing DNA in this way can fix broken genes or change cellular functions.
Which type of repair will happen?
Controlling which type of repair happens after Cas9 makes a cut can be difficult. Small indels made via NHEJ are the most common genome-editing outcome. Adding in template DNA can lead to its insertion at the cut site, but generally not every time and not in every cell the researcher wants to edit. In a single experiment, it would be unsurprising to find only 1 out of 1000 cells with the right insertion! DSBs can also lead to big, unwanted rearrangements or deletions in chromosomes in a small percentage of cells, called on-target effects.
So what do scientists do? In some cases, researchers can isolate just the cells or organisms that received the edits they want. Sometimes, they add in small molecules or design their template DNA in a way that encourages the cells to use the HDR pathway. Scientists are also developing variations of CRISPR genome editing that lead to more reliable repair outcomes, like ‘base editors’ described later on in "Expanding the CRISPR toolkit." The best strategy depends on exactly what the researcher is trying to do, and there’s room for more innovation.
🎥 | We've gone through the molecular details of genome editing, but what does the real-life process look like for someone performing the procedure in a lab? Peek inside one of our labs and see how genome editing is done in the video below.
Refining CRISPR tools
Making genome editing more accurate and more versatile
- Vocabulary
- Nickase
Where is there room for improvement?
Genome-editing experiments rely on CRISPR tools to cut specific DNA sites, leading to changes in the DNA sequence. CRISPR tools make it very easy to perform genome-editing experiments, but they have important technical limitations:
- They don’t always cut at the correct site in the genome
- There are some limits to which sites they can target
As you’ll learn below, researchers are finding ways to overcome these challenges.
Making genome editing more accurate
CRISPR tools don’t always cut the correct DNA sequences. Sometimes, if there’s a partial match between the guide RNA and target DNA, it’s enough for the enzyme to bind and cut. A CRISPR protein cutting at an unwanted site is called an off-target effect. Genome editors typically only want to edit the target they’ve selected, so off-target edits can cause problems.
Recognizing this issue, researchers have modified CRISPR tools by altering their structures. These enzymes are better at obeying their guides and rarely cut the wrong sequences. It’s important to check for off-target edits no matter which CRISPR tool you use, but using these “high-fidelity” variants generally improves accuracy. There are other methods to improve accuracy as well, sometimes by modifying the guide RNA instead of tweaking a Cas protein.
CRISPR nickases make genome editing more accurate in a different way. CRISPR enzymes like Cas9 usually cut both of DNA’s two strands. Researchers can mutate one of Cas9’s cutting domains to stop it from working, turning the enzyme into a nickase. A nickase makes a cut in just one strand, which is called a nick. Individual nicks don’t typically lead to genome editing on their own, but two nearby nicks do. Scientists can load CRISPR nickases with two different guide RNAs, targeting them to DNA sites that are close together and triggering genome editing.
Why would this type of genome editing be more accurate? Think of cutting DNA sequences with CRISPR tools like shooting arrows at targets. Even a super skilled archer will occasionally miss a target. However, it is unlikely that a skilled archer will miss the target in the same way twice. Similarly, it is unlikely that two CRISPR nickases will create off-target nicks in the same area of the genome. Off-target cut sites can be anywhere throughout the genome, so they’re unlikely to happen right next to each other. Unless there are two nicks close together, no editing will happen. So, off-target editing using dual nickases is rare.
Making new DNA sequences editable with PAM variants
Common CRISPR tools can target many DNA sequences. However, CRISPR tools generally require their targets to have licensing sequences called PAMs, explained in depth in the previous chapter. PAMs are typically short (sometimes just two nucleotides), so a scientist can usually find one near the site in the genome that they’d like to edit. But not always, and they might not be able to find a PAM that will allow the CRISPR enzyme to cut exactly where they’d like make an edit. So, the PAM requirement limits the number of targetable DNA sequences.
Researchers are getting around these limitations by discovering new CRISPR enzymes or engineering their existing tools. They can digitally search through DNA sequences and find CRISPR systems with different PAM requirements. They can also change a protein’s PAM requirement. To do so, they modify the amino acids in the protein that interact with the PAM. With current techniques, researchers can target essentially any DNA sequence!
Expanding the CRISPR toolkit
CRISPR's programmable DNA targeting can be combined with other useful functions
- Vocabulary
- dCas9, fusion, base editors, transcribe/transcription, gene regulation, activator, repressor, knock down, recruit, epigenome
Turning Cas9 into a swiss army knife for researchers
During CRISPR genome editing, researchers generally cut a specific DNA sequence to make an edit. This technique is used for many exciting research projects. Nonetheless, the CRISPR toolkit has many tools that do much more than cut DNA. New methods are invented often, so we describe just a handful of the key tools in this section.
The majority of these tools use a non-cutting CRISPR protein called "dead" Cas9, or dCas9, for short. If the common CRISPR cutting tools are like precisely targeted arrows, dCas9 is like an arrow with a blunted tip. dCas9 can still target precise DNA sequences, but it doesn’t cut them.
It’s possible to attach proteins with various functions to dCas9. This is usually done by making a physical attachment called a fusion. The fused proteins keep their original abilities and gain dCas9’s ability to find and latch onto specific DNA targets. This is like tying a payload to a blunted arrow. Researchers use these tools to target protein activities to specific DNA sequences.
Editing DNA directly with base editors
Base editors are variations of the CRISPR-Cas9 system that directly change DNA bases. There are naturally-occurring and engineered enzymes that can convert individual letters in a DNA sequence into other letters. When fused to Cas9, these enzymes can be targeted to a specific DNA sequence. Base editors usually include dCas9 or Cas9 nickase. This means there are no double-strand breaks, and base editing does not activate the repair systems triggered by traditional genome editing.
So, scientists can use base editors to precisely change one DNA letter to another. This process is like highlighting a single letter in a text document and replacing it in one keystroke. Right now, the main base-editing tools let researchers change A to G and C to T. There has been substantial progress in engineering new and improved base editors, and more of these tools may become available soon.
Turning gene expression up and down
Genes encode proteins. To create an encoded protein, the cell first copies or transcribes a particular gene into mRNA. The mRNA is then used like an instruction sheet that tells ribosomes how to build the protein. Generally, if more transcription occurs, then more of the encoded protein is created. If there is less transcription of mRNA, less of the protein product of that mRNA is made.
In order to function properly, grow, and reproduce, cells constantly need to adjust their levels of individual proteins. This is called gene regulation. To do this, regulator proteins in the cell can turn transcription of a gene up or down, generating more or fewer mRNA copies and more or less of the encoded protein product. A regulator protein that increases transcription is called an activator, and a protein that decreases transcription is a repressor.
Researchers can attach one ore more of these transcriptional regulators to dCas9 and program it with a guide RNA targeting a gene of interest. When the dCas9 binds to its target, the regulator turns transcription of the gene up or down. In this way, scientists can learn more about the gene’s function.
This approach is especially useful if fully knocking out, or breaking, a gene is lethal to cells. Researchers can instead reduce gene expression, called knocking down the gene, which usually lets the cells survive while still providing clues about what the gene does.
How do dCas9–regulators actually control transcription? In some cases, dCas9 is thought to physically block proteins like RNA polymerase from binding near genes, preventing transcription. Sometimes, the regulator proteins attract or recruit other proteins that affect transcription. This can also be done by directly modifying the epigenome with CRISPR tools, explained in the next subsection.
Making epigenetic modifications
All the DNA in a cell is called the genome. The epigenome is a set of chemical modifications on the DNA and the special proteins around which DNA is wrapped, histones. These chemical marks can affect transcription, activating or repressing genes. If we imagine the genome is a cookbook and genes are recipes, making an epigenetic mark is like adding a sticky note to a recipe that says “Make more of this!” or “Make less!”
The effects of epigenetic modifications can be complex, but researchers understand many of the proteins that make them. By attaching epigenome-modifying proteins to dCas9, scientists can target them to specific genes to turn expression “up” or “down.” They typically target them just before the start of a gene, in a region called the promoter, where transcription machinery has to attach before transcribing a gene into mRNA.
Some of the most common epigenetic editing tools consist of dCas9 fused to a methyltransferase enzyme, which adds methyl groups to DNA. DNA methylation can partially or fully prevent transcription. dCas9–methyltransferases thereby turn genes “down” or “off.”
Conversely, researchers can activate genes using dCas9–demethylases, which remove methyl groups. dCas9 can also be fused to an acetylase, an enzyme that adds acetyl groups to a part of the histone called the tail. These acetyl marks tend to “open up” DNA and increase transcription.
Direct epigenetic edits tend to turn genes more completely and permanently “on” or “off” than the CRISPR regulators described in the previous subsection, but it depends on the experimental design. Scientists can use epigenetic CRISPR editors to learn about the complex consequences of epigenetic modifications.
Making DNA sequences glow
There are many types of proteins and small chemical dyes that emit light. They come in all colors of the rainbow. One of the most well-known fluorescent proteins is green fluorescent protein, or GFP.
Researchers can fuse a fluorescent protein or dye to dCas9. Targeting a fluorescently-labeled dCas9 to a specific DNA sequence will make it light up. Researchers can then use special microscopes to track how these sequences interact with other parts of the cell, detect when other fluorescent proteins bind at the same site, and more. Interactions like these can influence important cellular functions.
🎥 | To review the basic genome-editing process and watch how other tools in the CRISPR toolkit work, check out the animation below.
Using CRISPR for basic research
By altering genes and seeing what happens, we can learn about what those genes do
- Vocabulary
- Basic research, hypothesize, hypothesis, gene expression, gene repression, CRISPR activation/CRISPRa, CRISPR inhibition/CRISPRi, model organism
Learning fundamental facts about biology through experimentation
Scientists can ask a huge variety of questions about the world, and not all research is designed to solve a problem. Basic research involves exploring fundamental aspects of nature and biology, and is often motivated by curiosity, awe, and fascination.
Researchers use CRISPR tools to answer many basic research questions about how genes and genomes influence how cells and organisms work. This style of experimentation typically involves making changes to genes and looking for effects on the cell or organism. Tweaking genes has been possible for many years, but CRISPR speeds up the process dramatically and can enable modifications in organisms that haven't been studied before. The types of gene modifications that basic research scientists make fall into four major categories:
- Knocking out genes
- Turning the effects of genes up or down
- Changing gene functions
- Adding new genes
The subsections below show how researchers use these modification techniques to learn about genes. Each subsection contains two examples. The first example shows how a mechanic could use a similar technique to learn how a bike functions (denoted with 🚲). The second example shows how a biology researcher could use the technique to learn how a plant gene works (marked with 🌺).
Breaking genes to learn about what they do
With CRISPR-based genome editing, researchers can delete or knock out specific genes. By removing or messing up key parts of a gene's sequence, they "break" it, or stop it from functioning. By monitoring the effects of breaking the gene, they get clues about what the gene normally does. For example, breaking a gene might change an observable trait. One could then make an informed guess or hypothesize that the gene controls that trait. Further experiments will refine this hypothesis and build understanding.
🚲 | A mechanic could use a similar technique to learn how a bike works. The mechanic starts by destroying the bike’s brakes. The mechanic then has a volunteer ride the bike. As a result, the volunteer has difficulty stopping. So, the mechanic concludes that brakes likely enable riders to stop. The mechanic is making a hypothesis — an idea about how things might work based on an observation. A hypothesis can be tested with more experiments, which will support or disprove it.
🌺 | Similarly, biology researchers learn what plant genes do. The researchers start by using CRISPR genome editing to break a specific gene. Later, they notice that the plant’s flower petals turn from blue to white. So, they hypothesize that that the gene controls flower color. This is a good starting point for future experiments!
Turning a gene's effect "up" or "down"
CRISPR tools can be used to activate or "turn up" gene function, like turning up the volume on a TV. Remember that genes are the instructions for proteins, so activating a gene means telling the cell to make more of the protein that the gene encodes. This is known as increasing the gene's expression. Genes can also be "turned down," so the cell makes less of the protein they encode. This is called gene repression. Two CRISPR methods used to do this are CRISPR activation (CRISPRa, turning up expression) and CRISPR inhibition (CRISPRi, turning down expression).
Researchers turn genes up and down to test hypotheses about what they do.
🚲 | The mechanic can use a similar technique to learn more about brakes. The mechanic can “turn up” the brakes by adding more to a bike. With extra brakes, the mechanic hypothesizes that a rider will stop more easily.
The mechanic adds more brakes to a bike and watches a volunteer ride. The volunteer stops more quickly when they brake. Brakes help bikes stop.
🌺 | The biology researchers know their gene impacts flower color. They hypothesize that turning up the gene function will make their flower more colorful. They use CRISPR tools to turn up the gene's expression and the plant’s flowers turn a deeper blue. This result provides supporting evidence for their original hypothesis, that the gene controls flower color. It also lets the researchers refine their hypothesis, making it more specific. They now believe that the gene controls the production of a blue pigment. They conclude that turning up the gene's function means more pigment gets made, making the flowers a darker blue.
Using CRISPR tools to change gene function
Editing the sequence of a gene with CRISPR can change how the gene works or what it does. It's hard to design edits that will have a specific effect on gene function without already having a pretty good idea of how the gene works. This information usually comes from previous experiments and observations.
🚲 | Similarly, the mechanic can alter the brakes to learn more about them. The mechanic knows that brakes help riders stop and notices that the brakes are connected to the bike’s back wheel. The mechanic is not sure if this connection is important. The mechanic thinks it’s probably only important for the brakes to connect to a wheel. The mechanic hypothesizes that if they switch the brakes to be connected to the front wheel, the bike will still stop.
The mechanic connects the brakes to the front wheel. Afterwards, a rider can still stop. This result provides evidence that supports the mechanic’s hypothesis. Brakes can be connected to either the front or the back wheel and the bike will still stop.
🌺 | The biologists know about a gene involved in leaf color that has a similar sequence to the gene that they've linked to flower color. Upon close examination, they notice that the beginning of the genes are quite similar, but the ends are very different. The flowers and leaves are different colors, so they hypothesize that the ends of these genes are the parts that control color.
To test this hypothesis, the researchers replace the end of the "flower color gene" with the end of the “leaf color gene." As a result, the flower petals turn green! This result provides evidence for their hypothesis. The "flower color gene" is important for giving the flower color, and the end of the gene controls which color develops.
Using CRISPR tools to add new genes to organisms
Last, researchers can use CRISPR technologies to insert entire new genes into organisms. When given a new gene, an organism may gain an ability. This is good evidence that the gene controls the ability.
🚲 | The mechanic can learn even more about brakes by adding them onto a different vehicle. The mechanic has a scooter and knows scooters very well. This particular scooter has no stopping mechanism. The mechanic therefore hypothesizes that adding brakes to the scooter will enable a rider to stop. A volunteer who rides the modified scooter is able to stop. This result provides evidence for the mechanic’s hypothesis. Brakes enable riders to stop.
🌺 | Biology researchers similarly need to control their experimental conditions. The first three plant experiments were controlled. In these experiments, researchers directly compared plants with and without genomic changes. They only saw one change each time. As a result, researchers could attribute the observed changes to the genomic changes they made with CRISPR genome editing.
When adding a new gene to an organism, researchers need to know what effects they're looking for. Then they can choose an organism where these effects will be easy to measure. For this reason, researchers often insert new genes into model organisms. These are well-studied organisms whose growth and biology are well understood. In these organisms, it’s easy spot changes in biological activity.
For example, the plant researchers want to insert their “flower color gene” into a model plant. They hypothesize that this will turn the new plant's flowers blue. It would be a bad idea to insert this gene into plants with blue flowers or no flowers. It would be very hard to observe any effects in these plants!
So, the researchers insert the flower color gene into a plant with red flowers. They hypothesize that the petals will turn purple because blue and red pigments will both be produced. But, they observe that the petals actually turn blue. This provides even more evidence that the gene controls flower color, and reveals something new — the "blue" gene from the original plant seems to "take over" in the red-flowered model plant, turning the petals totally blue instead of adding a blue hue to the red to make purple. This could be an interesting observation to explore in future experiments.
Refining hypotheses and improving understanding with CRISPR
Note how the results from these experiments build on each other. The researchers start with little understanding. Initial “breaking” (knockout) experiments provide broad clues about how things work. Later experiments ask more specific questions that come from these broad clues. In this way, hypotheses about gene function are refined.
Researchers uncover results that gradually improve understanding. Doing experiments to test hypotheses often leads to unexpected observations that give scientists new ideas to explore next. Eventually they’ll understand enough to predict how genetic modifications will change the biology of an organism. In the past, this was a very time-consuming process, but CRISPR technologies speed everything up. We're learning more and more how genes and organisms work.
CRISPR screens
Learning about many parts of the genome, all in one big experiment
- Vocabulary
- Genetic screen, genome-wide screen, pooled screen, pooled library, arrayed screen, positive selection, negative selection, reporter
Using CRISPR to link genes to traits
More and more genomes are being sequenced over time. Yet even in extensively-studied organisms, we still don’t know what all genes do or how certain DNA sequences help control gene expression. Using CRISPR tools, we can edit genes to learn about the proteins they encode. It’s easy for researchers to edit single genes using CRISPR tools. It is much more difficult to identify unknown genes responsible for cellular traits. Researchers could edit and test every gene one-by-one — but that would take a lot of time.
Genetic screens offer a faster alternative. Researchers use CRISPR tools to screen many genes in a single experiment. The word “screen” means to search through or test systematically. When applied to CRISPR technologies, screens allow researchers to systematically test genes.
Pooled CRISPR screens allow researchers to test tens of thousands of genetic targets at once. Because so many DNA targets can be modified in one experiment, including all ~20,000–25,000 genes in the human genome, these are often referred to as genome-wide screens.
In a pooled CRISPR screen, researchers deliver CRISPR proteins plus a pooled library of guide RNAs (gRNAs) to a large batch of cells. Just like a real library contains many different books, a gRNA library is a mixture of many gRNAs — the small, customizable molecules that guide CRISPR proteins like Cas9 to specific DNA targets.
Researchers add just the right amount of Cas9 and guide RNAs (usually these components are added by introducing DNA that encodes them) to the mix to ensure that, on average, only one gRNA ends up inside each cell. The aim is to see the effect of modifying one gene at a time, rather than the complicated effects that might occur when CRISPR enzymes alter multiple genes in a single cell.
Researchers then grow all the cells under specific conditions that only allow cells exhibiting a certain trait to survive. This process of deliberately killing off a subset of cells in a mixed population is called 'selection.' The selection reveals whether the CRISPR modification affects a particular trait. Researchers then identify the targeted genes in affected cells by sequencing the remaining gRNAs (or the DNA that encodes the gRNAs). In the cells that died, all the RNA and DNA is destroyed and won’t show up in the sequencing results. The gRNA sequences that do show up must have come from surviving cells, and researchers can look to see which genes those gRNAs targeted. These genes likely have some biological link to the trait of interest. They may even directly cause it. We’ll cover selection and analysis in more detail in the next subsections.
Unlike in a pooled screen, in an arrayed CRISPR screen, genes are targeted one-by-one, but many are tested in parallel. Cells are kept in individual ‘wells’ of a multi-well plate and one gene target is tested in each well — basically, instead of a single dish of a cells, researchers use a dish with multiple separate cups inside, almost like dozens of individual test tubes all taped together into a single block. This lets them do many experiments on individual DNA targets in parallel. Arrayed CRISPR screens are used less often and are generally limited to testing dozens or hundreds of targets at once, so we focus on describing genome-wide, pooled screens in this section.
CRISPR screens can involve making direct edits to DNA, like knocking out a gene. They can also make more subtle modifications, like turning down gene expression, described in “Expanding the CRISPR toolkit” above. Sometimes researchers don’t even want to target genes directly — they may instead use a CRISPR screen to study ‘non-coding’ sequences in the genome, which do not encode proteins and are sometimes misleadingly referred to as "junk DNA." This can help reveal things like how genes are regulated.
Selection and analyzing the results of a CRISPR screen
Researchers need easy ways to examine cells modified in pooled CRISPR screens. We can think of these researchers as gold prospectors. They must determine if a mine (experiment) has gold (targeted genes that affect the trait of interest). They also want to limit the amount of dirt (cells) they have to sift through.
Selection is a great way to analyze large numbers of cells. Selections come in two flavors:
In a positive selection, cells die if the CRISPR-based modification does not affect the trait of interest. Cells live when the target modification does affect the trait of interest. Researchers only need to examine the surviving cells to find genes that impact the trait of interest. This is like a gold prospector getting a map of all the gold-filled mining sites in the area.
For example, let’s say researchers want to find genes that increase bacterial antibiotic resistance. They can use CRISPR tools to activate many different genes in a batch of bacterial cells, where each cell has just one gene activated by a CRISPR tool. For a positive selection, they could attempt to grow the bacteria on a petri dish with an antibiotic. Normally, all the bacteria would die. Bacteria that grow are able to resist the antibiotic. This means that genes activated in these surviving bacteria likely increase antibiotic resistance. Follow-up experiments could confirm this and uncover how these genes increase antibiotic resistance.
In a negative selection, cells die if the CRISPR-based modification affects the trait of interest. Cells live when the target modification does not affect the trait of interest. But you can’t analyze dead cells, so how can you tell which genes were targeted in cells that are now gone? Researchers look to see which guide RNA sequences are still present, and work backward in a multi-step process:
- First they create a list of all the genes that they targeted with the library of gRNAs.
- Then they create a list of all the genes targeted in the surviving cells.
- Finally, they subtract the second list from the first list. Genes targeted in the dead cells remain. These genes likely impact the trait.
This is like a gold prospector getting a list of all mining sites that lack gold. The prospector can use this list to determine which sites do have gold. They just need to reference another list of all mining sites first. This takes an extra step, but it’s still quicker than examining every mine individually.
For example, let’s say researchers want to find genes that let bacteria eat a particular kind of sugar. They can use a CRISPR protein and a gRNA library to inactivate many different genes across a culture of bacterial cells, where each cell has just one gene repressed or knocked out. For a negative selection, they could attempt to grow the bacteria in a dish with the sugar as the only food source. Most bacteria will survive, but those that die were no longer able to use that sugar as a food source. This means that the genes that were inactivated in those bacteria are likely involved in digesting the sugar. Follow-up experiments could validate these findings and uncover how these genes enable bacteria to eat the sugar.
Sometimes selection is not possible, but researchers may have quick ways to examine every modified cell. In these cases, CRISPR screens are still feasible. For example, cells might fluoresce, or “glow,” if gene targeting affects the trait of interest. This might be done using a reporter molecule like green fluorescent protein. Researchers can use various types of lab equipment to isolate glowing cells. They can then identify the targeted genes in these cells. These genes likely impact the trait of interest. This would be like a gold prospector using a grate to sift gold from dirt. The prospector doesn’t have to examine all the dirt by hand, but does have to run it all through the grate.
In the worst case, researchers have to tediously examine every single cell. This could happen if they are studying a trait needing microscopic study. Such examination would be impractical with large gRNA libraries. It would be like the gold prospector sifting through all the dirt in a mine by hand. It’s practical only if there's just a small amount of dirt.
An Example CRISPR Screen in Bacteria
Let's run through a hypothetical CRISPR screen, illustrated in detail below.
Imagine that researchers would like to find genes that let bacteria grow by eating a sugar called aspartame. They can use a pool of guide RNAs to repress (turn down transcription of) many different genes in bacteria, repressing one gene per bacterial cell.
Next, the researchers transfer the bacteria to a growth medium containing aspartame as the only food source. Bacteria containing gRNAs that repress genes important for growth on aspartame should not grow, so this is negative selection.
By identifying all the gRNAs remaining in the bacteria that do grow on aspartame, researchers can also determine which gRNAs are missing. By later checking the genes targeted by these gRNAs, they can create a list of candidate genes that may be important for aspartame digestion. These genes can be retested individually and explored more in future experiments.
Studies enabled by CRISPR screens
Researchers use CRISPR screens for many reasons. Some of their recent uses include identifying human genes that:
- ...impact HIV infection. Understanding how human genes impact HIV infection might enable new treatments. Learn more >
- ...are important for Chlamydia trachomatis infection. C. trachomatis is a bacterium. It is a leading cause of sexually transmitted infections. Understanding C. trachomatis infection may lead to new and improved treatments. Learn more >
- ...cancer cells rely on. Cancers like acute myeloid leukemia are treated with chemotherapy, but it doesn't always work. If we understand the genes that are important for cancer to grow and spread, we might be able to design new drugs that target those genes. Learn more >
Gene drives
- Vocabulary
- Gene drive
Using CRISPR to break the rules of classical genetics
Imagine a teenager cornering one of her parents and asking for advice on something. Next, she asks her other parent for advice on the same thing. In some cases, their advice will be the same, but often she’ll get two different pieces of advice.
Genes work in a similar way. You have two complete sets of genes – one from each biological parent. Sometimes the gene pairs are identical, but you often end up with two different versions of the same gene. Your specific mixture of genes is part of what makes you who you are.
Now imagine that the teen sits down with her parents and asks for advice from both at the same time. In some cases, they'll agree. Other times, one parent will override the other and the daughter will have to take only their advice.
Gene drives work like this latter scenario. They break the normal rules of genetics. If an organism inherits a gene drive from one biological parent and a normal gene from the other, the gene drive will override and replace the normal gene. They are guaranteed to pass the gene drive onto their offspring, where the replacement process will repeat.
Scientists can use CRISPR tools to create gene drives. Gene drives spread chosen genes through populations of sexually-reproducing organisms with short lifespans. For example, scientists could potentially use gene drives to spread a gene through many, many, many mosquitoes to stop them from transmitting malaria. While gene drives would not work well in humans, they have the potential to improve human well-being. However, their potential for broad effects comes with possible ecological impacts and ethical questions. Developers and affected communities must consider these impacts before deploying gene drives.
CRISPR gene drives
There are natural gene drives, but these are tough to redesign. CRISPR tools let researchers make new gene drives with custom purposes. CRISPR gene drives work in the following way:
- Researchers generate the initial organisms carrying the gene drive, which includes a CRISPR nuclease gene, a guide RNA, and usually a "cargo" or "payload" gene. The cargo can be a foreign gene, but is most often an edited version of one of the organism's naturally-occurring genes.
- The researchers let the gene drive organisms mate or breed with unmodified organisms. The offspring will therefore receive a chromosome that carries the gene drive (from the gene drive mosquito) and the other copy of the chromosome (from the unmodified mosquito) will carry the normal version of the gene.
- The CRISPR nuclease and guide RNA are produced and they assemble together. The guide RNA directs the CRISPR protein to cut at the desired target site, which is usually the unaltered version of the cargo gene.
- The cell repairs the cut by pasting in the entire gene drive, because the DNA surrounding the gene drive components is homologous to the sequences at the cut site (for an overview of homology-directed repair, read the "CRISPR genome editing" section). As a result, the gene drive replaces the other versions of the cargo gene. The offspring will now carry the gene drive on both copies of the chromosome.
- Future offspring inherit the gene drive and the replacement process repeats. The cargo gene is passed down in every generation, spreading rapidly through the population.
Uses for gene drives and their limitations
Gene drives can spread desired genes or traits to all members of a population. For example, gene drives might rid all mosquitoes of the parasite that causes malaria. Or, they might make a certain weed susceptible to herbicides. They might even make dangerous species sterile, causing eventual extinction.
These applications are exciting, but they have two key limitations:
- Gene drives only spread through organisms that reproduce sexually. They are ineffective in many types of bacteria and other asexual organisms.
- Gene drives only spread to future generations. So, population-level applications are practical in organisms with short lifespans, but not organisms with long lifespans.
Gene drives have the potential for huge societal impacts
If deployed, gene drives could change entire species. They would also impact human societies that live alongside species, use them for food or practical purposes, or revere them. Further, impacting one species can have unforeseeable ripple effects on whole ecosystems.
In order to use gene drives ethically, researchers must communicate deployment risks and benefits to societies who would be affected. Theses societies should then decide whether to deploy gene drives.
In some use cases, the affected society could encompass multiple countries, whole continents, or even the entire world, depending on where a certain species is found. Such a diverse society is unlikely to reach a consensus, so planet-scale applications of gene drives are unlikely.
Responsible deployment of a gene drive might be more feasible in a small, controlled setting. For example, researchers might deploy gene drives to eradicate pests on small islands. In these situations, the chances for unexpected gene drive spread are very small. As such, the local society could potentially provide consent for gene drive deployment.
Gene drive control mechanisms
Some concerns surrounding gene drives include:
- Moral, philosophical, or spiritual objections to genome editing
- Unanticipated ecological effects
- Spread beyond the intended ecosystem
Researchers must meet the first concern with open dialogue, looking to share scientific understanding rather than impose personal opinions. Researchers can address the latter two concerns using control mechanisms. These put “brakes” on gene drive spread.
Gene drive control mechanisms include:
- Limiting gene drive inheritance - Researchers are developing “daisy-chain gene drives.” These use limited "genetic fuel" to spread through a small number of generations. They can treat problems restricted to small geographical locations.
- Creating barriers to inheritance - It’s possible to genetically modify an organism so a gene drive won’t affect it. Organisms that are “immunized” against gene drives can act as barriers to gene drive spread.
- Overriding one gene drive with another - A gene drive might cause unanticipated issues. In this case, researchers may be able release a new "reversal" gene drive that could destroy the original gene drive.
With these mechanisms, we have greater ability to control gene drives to respond to unanticipated or unwanted outcomes of gene drives. Note that these controls still function through large-scale genetic modification, so they would also need societal approval to be deployed.
So far, gene drives have been treated with extreme caution. It is not clear that gene drives will ever be used outside of research settings.
