Imaging unique DNA sequences in individual cells using a CRISPR-Cas9-based, split luciferase biosensor
Heath NG, O'Geen H, Halmai NB, Corn JE, and Segal DJ. Frontiers in Genome Editing
(2022)

We are developing methods to detect specific genome edits in individual living cells, overcoming an historic bottleneck that will allow us to edit difficult cells much faster.
SHARE:
With the advent of CRISPR-Cas9, the primary bottleneck in genome editing is no longer the nuclease. Among the remaining challenges is the ability to identify and isolate cells in which the desired genetic or epigenetic events have occurred. This is of particular concern for cell types or procedures in which the frequency of successful genome edits is low, such as homology directed repair (HDR) in primary cells and stem cells. Indeed, a considerable portion of the time required for genome editing is often the isolation of cells with the desired genotype.
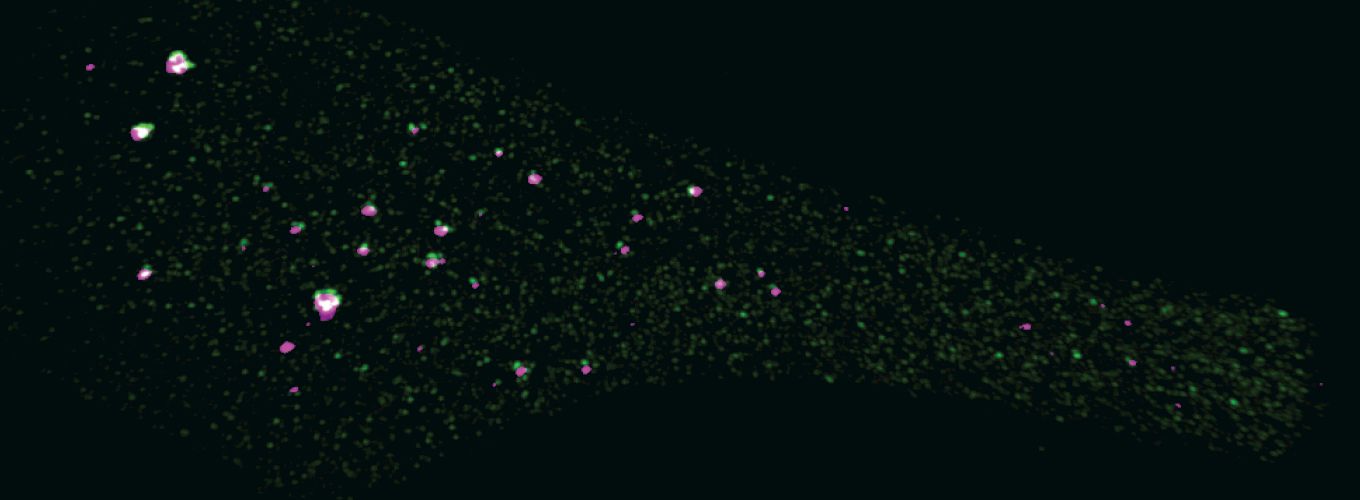
Tethering fluorescent proteins to DNA-binding proteins, including catalytically dead Cas9 (dCas9), creates molecular probes of specific DNA sequences in living cells. However, it is important that such probes are “always on,” meaning that it is impossible to distinguish between a probe bound to a target site from one floating free in the nucleus. For that reason, the use of such probes has been limited to regions containing tandemly repeated sequences or using 26–37 gRNAs. Such a system would not be useful to detect unique DNA edits.
The scientific premise of this study is that the use of “turn-on” probes can reduce the number of tandem binding sites in the genome to one unique site, allowing interrogation of the sequence of an individual genomic locus. In principle, one could apply such probes to a pool of treated cells, then use fluorescent microscopy, flow cytometry, or a similar detection system to isolate specifically modified cells. The use of split-reporters to detect genetic changes in living cells could potentially impact many users around the world. The outcome of these experiments could be a transformative step forward in the efficiency of genome editing.
Share this project: