New FDA guidance for platform therapies
Victoria Gray was born with a genetic mutation that causes sickle cell disease. Just one DNA letter change in her genetic code created a faulty protein, leading to years of fatigue, episodes of sudden, intense pain, and long, frequent trips to the hospital. In 2019, Gray became the first person with sickle cell disease to be treated in a clinical trial using CRISPR to restore the faulty protein to its healthy form. Since then, her symptoms have decreased dramatically – no more hospitalizations, blood transfusions, or missing out on special moments with her family.
“This is really a life-changer for me. It’s magnificent,” Gray told NPR. In 2023, her treatment was approved by the FDA under the name Casgevy, with approvals following in the EU and UK.

“At this point, all hypotheticals, the words ‘potentially’ and ‘could’ or ‘in principle’ are gone,” says Fyodor Urnov, IGI’s Director of Technology & Translation. “CRISPR is curative.”
Like Gray, over 200 million people worldwide are living with rare and neglected diseases that have a profound effect on their lives. Many of these diseases are caused by small changes in DNA sequences, often just a change in a single DNA letter. What will it take for the potential of CRISPR to turn into real therapies that are widely available?
The path to CRISPR cures for rare and neglected diseases
The potential for CRISPR cures creates hope, but also brings up challenges for scientists, drug makers, and regulators, especially when it comes to addressing rare diseases. How do you test a treatment if you don’t have enough patients for a clinical trial? How do you decide if a treatment is safe enough if only a handful of people — or just one person — has been diagnosed with the disease it could treat? How do you drive research forward when there are too few patients for profit-driven pharmaceutical and biotech companies to take an interest?
How treatments usually get to the doctor’s office
When scientists have an idea for a new treatment, they begin pre-clinical research, which involves lab experiments to make the right formulation and test the treatment in isolated human cells and lab animals. This process takes multiple years and, in the United States, it concludes when researchers submit their data and a plan for a human test called a clinical trial to the U.S. Food and Drug Administration (FDA). If the data and plans are sound, the FDA will let the researchers start testing if the treatment is safe and effective. Other regions of the world have their own versions of the process that follow similar steps.
Clinical trials are done in phases that each take multiple years. Phase 1 clinical trials usually include just a handful of people, and the goal is to make sure that the experimental treatment is safe. As phases progress, the number of patients enrolled increases, and in addition to safety, clinicians start assessing whether the treatment improves patient health. After the third or fourth phase, the FDA or similar governing bodies in other countries may approve the treatment for broad public use if the data show that it is safe and effective. From pre-clinical research to approval, developing a new therapy usually takes 10–15 years.
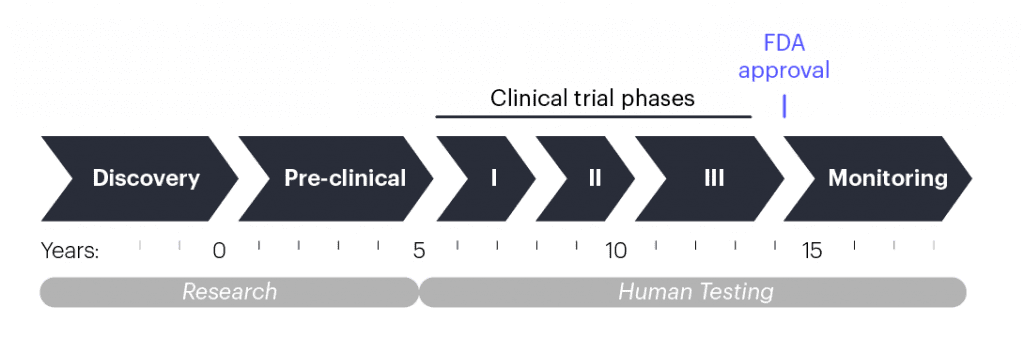
Clinical trial timelines
Rare and neglected diseases face additional hurdles. Financial incentives are the largest problem: developing a new disease treatment is incredibly expensive. Pharmaceutical and biotech companies take on these expenses based on predicted profit from a successful treatment, and the larger the patient group, the larger the potential profit. The smaller the patient group, the less chance to recoup research and development costs and make a profit.
“The fact that genome editing makes it possible to address all genetic diseases in principle doesn’t mean that biotech companies will take on every disease in practice. In the best case scenario, it takes three years to get to a clinical trial, which can cost more than $6 million per disease. Those are big hurdles for realizing the promise of CRISPR and for addressing unmet medical needs,” says Urnov.
The FDA developed the Orphan Drug Designation Program to create special financial incentives for developing treatments for rare (or “orphan”) diseases. This program has helped lead to the creation of 400 new therapies since its inception in 1983. However, 95% of the 5000 rare genetic diseases we know about still have no disease-modifying treatments. But new approaches are needed to advance the development of genomic therapies for rare diseases.
What’s different about gene-editing therapies?
Gene-editing therapies pose some unique technical challenges for both researchers and regulators.
- Delivery: Many researchers in the field consider delivery — figuring out how to get gene editing therapies to the right cells, and only those cells — the biggest technical challenge for gene-editing treatments. The IGI’s Delivery Collective was assembled to tackle this.
- Unknown side effects: There’s extra caution from researchers and regulators because the area is so new and the potential side effects of gene-editing treatments are still being uncovered. Researchers are focused on ensuring that gene editing is only occurring in the targeted areas of the genome, and that there are no potentially harmful immune system responses to the treatments.
- Lack of clinical diagnostic assays: Clinical diagnostic assays are needed to measure the effect of a treatment. For a gene-editing treatment, researchers might want to know what percentage of cells are edited or if there’s a change in the amount of a disease-causing protein. For rare diseases, these kinds of assessments usually don’t exist, and the cost of creating them is an obstacle. IGI’s Interventional Genomics Unit is currently developing clinical assays for blood cell disorders, including sickle-cell disease, as well as other rare diseases.
Platform technologies: a way forward for CRISPR cures
If you look at the numbers — 7000 rare diseases, 10–15 years for each therapy — the timeline seems impossibly long, even with institutions around the world tackling different diseases simultaneously. What can be done to accelerate the process?
One solution for rare genetic diseases could be a platform technology approach. A platform technology is when all the parts of a treatment are standardized into a single off-the-shelf package and only certain pieces of the platform change for a given disease. You can think of it like ordering a burrito: every burrito at your local shop might have the same tortilla, beans, rice, guacamole, and sour cream – this is the platform. The only thing that changes is the protein component that meets the needs of the person ordering the burrito: carnitas, grilled chicken, tofu, etc.
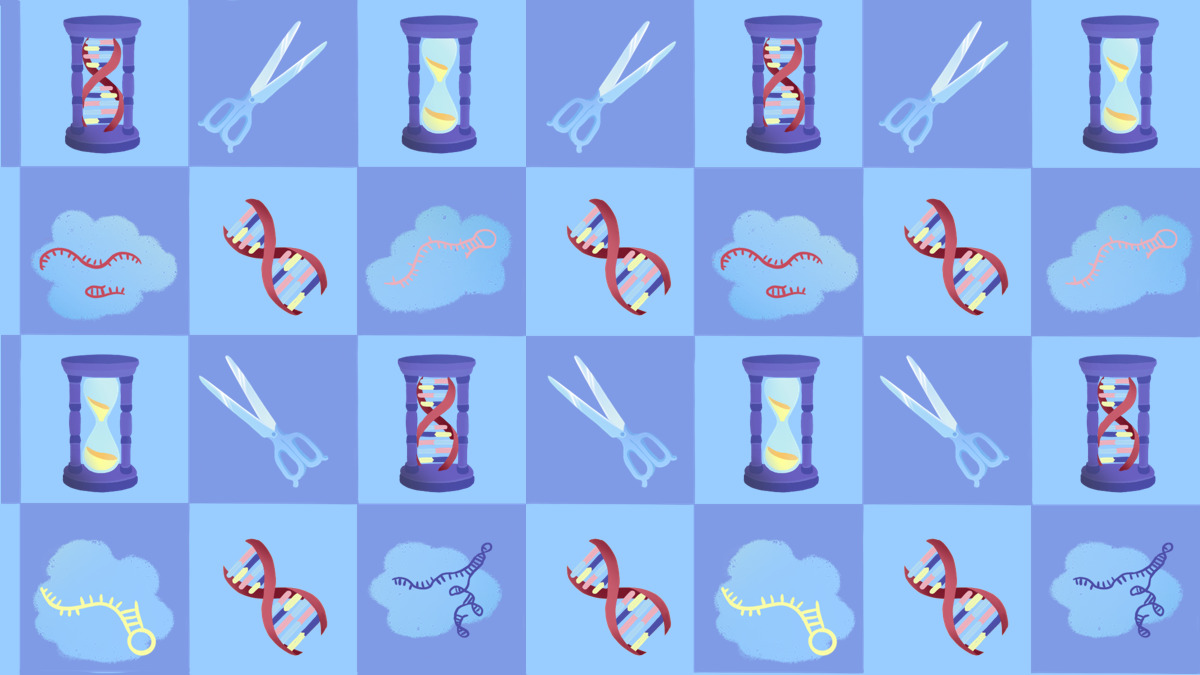
In CRISPR genome editing, a guide RNA molecule brings a Cas protein to a specific spot in the DNA, determined by the sequence of the guide. The Cas protein makes a cut in the DNA at that spot, and then DNA letters can be added, removed, or changed using a custom DNA template. So for CRISPR-based treatments, the basic burrito would be the Cas protein that cuts DNA, the method of delivery to the cells, how the treatment is administered, and what dose a patient gets. The only thing that would change would be the sequence of the guide RNA and any DNA repair templates that need to be specified for different edits.
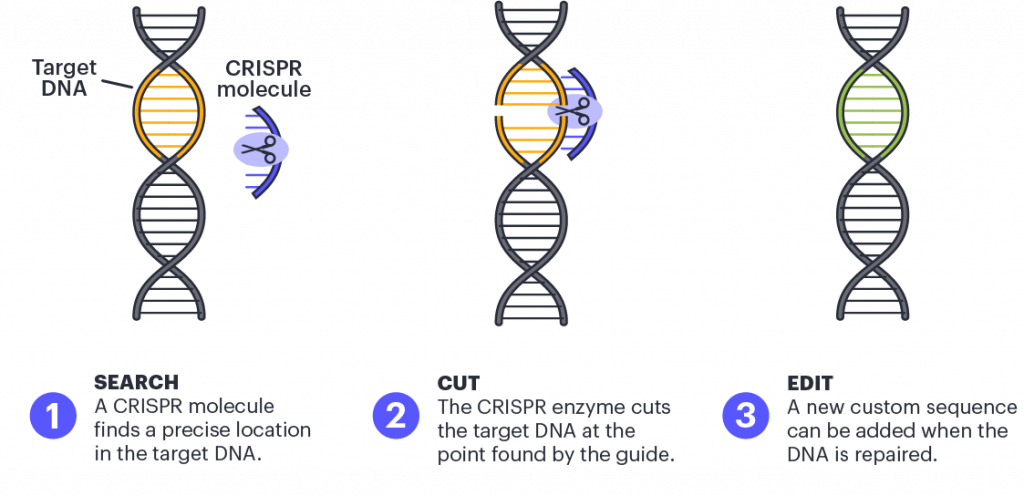
Scientists use CRISPR to find a precise location in the target DNA using a custom-made guide. A CRISPR-associated enzyme makes a precise cut in the target DNA. The cell repairs the break in its genome with the help of a new piece of DNA designed by the researcher.
Ultimately, the idea is that once the various parts of a platform are approved for one treatment, they wouldn’t need to be individually tested and approved again and again, dramatically cutting down on research, development, and clinical testing time, as well as on cost. For more common neglected diseases, this would dramatically lower the time and cost of the research needed to get a clinical trial started. For ultra-rare diseases, the FDA could use Emergency Use Authorizations to grant permission to use the platform technology to treat small groups of patients, or even a single patient.
New draft guidance from the FDA
In June 2024, the FDA released draft guidance for a new Platform Technology Designation Program, intended to safely reduce the cost of timeline for new cell and genome therapies. Under the proposed guidelines, researchers could submit certain data from already-approved products that use the same components as part of their application to initiate a clinical trial or get approval for therapy in trials. For example, if a company has successfully demonstrated the safety of a particular lipid nanoparticle for delivering CRISPR while testing a therapy for one condition, they could submit that data to the FDA to support the safety of using the same lipid nanoparticle, in the same concentration, for treating a different condition. This way, the company would not have to spend the money, effort, or time duplicating data.
Under the draft guidance, a therapeutic component could meet the platform technology designation if it is part of an already-approved therapy, if the existing safety data suggest that it could be used in other therapies without an adverse effect, and if the data indicates that the platform designation would make drug testing and approval significantly more efficient.
“We have known since Jennifer Doudna’s and Emmanuelle Charpentier’s 2012 Nobel-prize winning work that CRISPR is a platform for gene editing — swap out 20 nucleotides and you have a new experimental therapeutic! The central challenge now is to reduce this to clinical and regulatory practice — it’s the only way to get CRISPR cures to more patients who need them. This is a key goal for the IGI-Danaher Beacon for CRISPR Cures. We are planning to present our overall vision and a detailed plan to the FDA in the next year and have high hopes to benefit from the overall framing that the new Draft Guidance provides. Critically, we are committed to sharing our regulatory learnings broadly with the community of developers of gene-editing-based experimental therapies.”
Developing platforms at the IGI
Launched in January 2024, the IGI-Danaher Beacon for CRISPR Cures is aimed at speeding up the development of new genomic therapies using platform approaches. Creating new platform approaches for genetic diseases is also one of the main focuses of the IGI Center for Translational Genomics (CTG), housed on the first level of the IGI Building in Berkeley. Learn more about the CTG here. Both initiatives draw on the basic research expertise at UC Berkeley and clinical expertise at UCSF with the aim of helping patients with rare diagnoses.