Following up on my previous post about genome-wide CRISPR libraries, I thought it would be useful to show a bit more.
There are many things to consider when doing library work, but two major ones are
- How sure are you that a hit stems from on-target activity vs off-target trickery?
- What fraction of the library is functional?
On-vs-off target is the real worry, since you could spend a great deal of time chasing down spurious hits. CRISPR (and sh/siRNA) libraries tackle this problem with redundancy, and one should always require that a phenotype enrich multiple guides corresponding to the same gene. But in libraries with relatively low redundancy (e.g. GeCKOv1 only has 3-4 guides per gene), it’s easy to become enamored by a hit with a red-hot phenotype but only one guide.
The concern about functional fraction of the library is more technical, but impacts both ease of the screen and the redundancy point from above. If many of your guides are non-functional, all that extra work to clone and transduce your massive library vs a smaller one is wasted effort. Worse, your chance at redundancy is diminished with each non-functional guide.
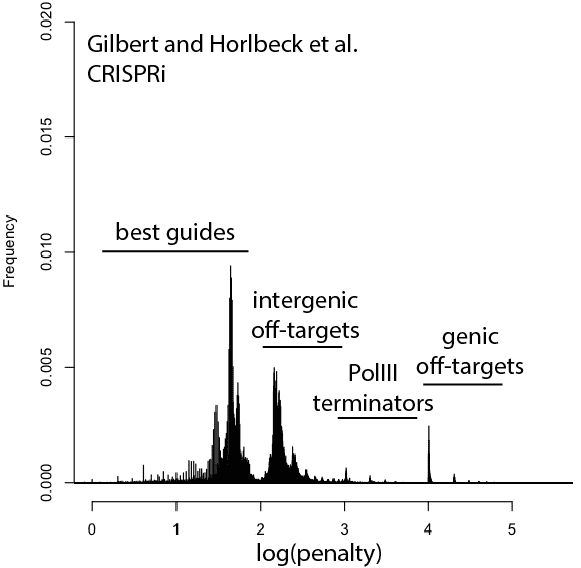
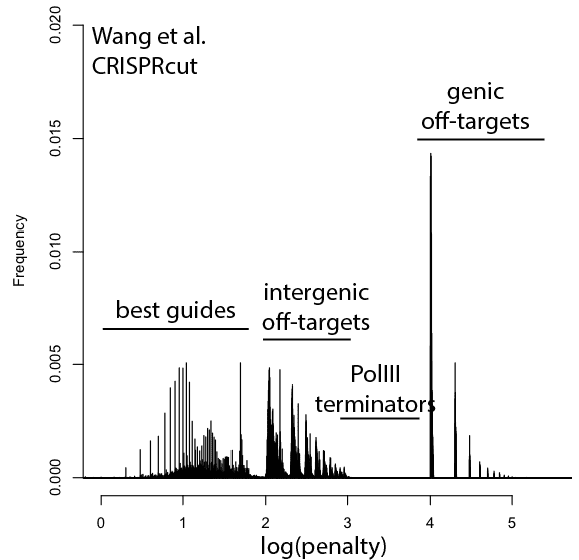
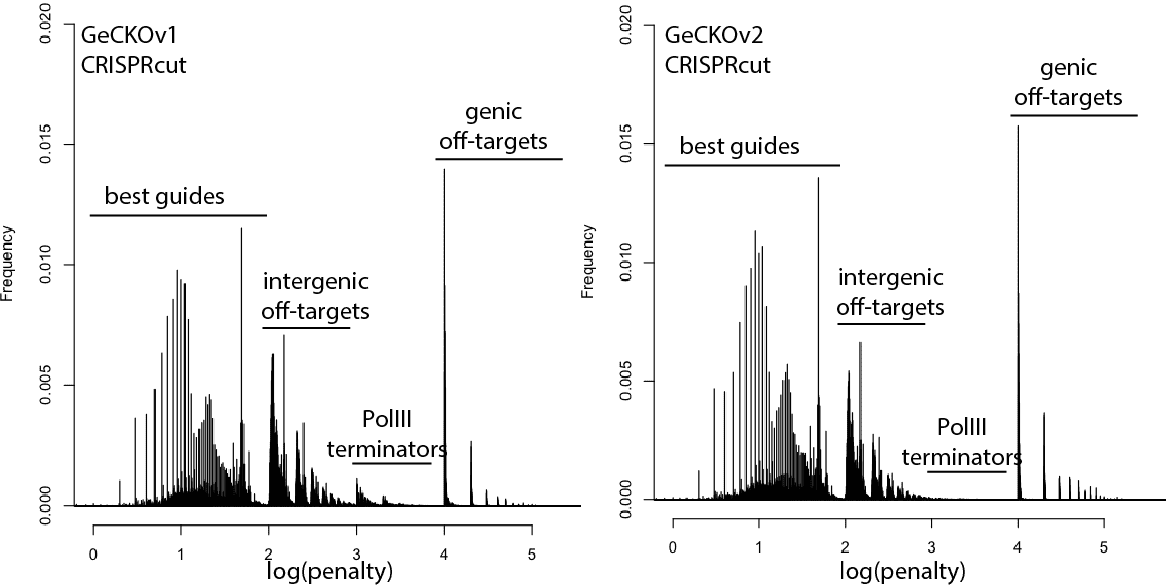
With that in mind, here are updated distributions for existing genome-wide guide libraries targeting human cells. The “penalty” axis is in log scale, and the penalties are easily interpretable to highlight the class of the problem. For penalties, the tens place represents the score of a guide itself, 100s place represents number of intergenic off-targets, and 1000s place represents genic offtargets.
For example, Anything with log(penalty)=0-2 has no off-targets and could be OK, though guides have much higher chance to be completely non-functional as one approaches 2. log(penalty)=2-3 have intergenic off-targets, with each 100-spike an additional off-target hit (e.g. penalty of 100 = one off-target, 200 = two off-targets, etc). log(penalty)=3-4 contain Pol III terminator sequences and are probably never even transcribed. log(penalty)=4+ have genic off-targets, with each 100-spike an additional off-target that impinges on a gene (e.g. penalty of 1000 = one off-target, 2000 = two off-targets). These penalties compound, so a score of 2,354 means two genic off-targets, 3 intergenic off-targets, and a guide penalty of 54.
Note that calling genic vs intergenic is done using Ensembl data and is sensitive to the type of CRISPR experiment. CRISPRi looks for hits within -50 to +300 of a gene, while CRISPRcutting looks at exons (for the moment we’ll leave aside the scary prospect of cutting within potentially functional intronic or UTR regions).
In general, things are looking pretty good for CRISPRi. There’s a bit of an advantage here, since CRISPRi only seems to work in a narrow window around the transcription start site, and so off-targets are less likely to hit a gene. CRISPRcutting libraries are not doing all that well with off-targets in annotated exons, and only deeper per-guide analysis would tell whether guide redundancy takes care of mis-called phenotypes. It’s nice to see that GeCKO has improved with v2 (e.g. got rid of terminator sequences), and hopefully v3 can get some of the genic off-targets under control.
I want to stress that all of these libraries work just fine and have been used successfully to give biological insight. But keep these guide properties in mind when working with each library and thinking about hits arising from their use.