My lab recently published a paper, together with outstanding co-corresponding authors David Martin (CHORI) and Dana Carroll (University of Utah), in which we used CRISPR to reverse the causative allele for sickle cell disease in bone marrow stem cells. This work got some press, so unlike other papers from the lab I won’t use the blog to explain what we did and found. You can go else where for that. But I do want to explain the motivation behind the work, as well as why we chose this approach.
Sickle cell disease and gene editing
Next-generation gene editing is already transforming the way scientists do research, but it also holds a great deal of promise for the cure of genetic diseases. One of the most most tractable genetic diseases for gene editing is sickle cell disease (SCD). The molecular basis has been known since 1949, so it’s relatively well understood. Its root cause is in bone marrow stem cells (aka hematopoietic stem cells, or “HSCs”), which are easy to get to with editing reagents. It’s monogenic and recessive, so you only need to reverse one disease allele for a cure. There’s no widely-used cure – though bone marrow transplantation from healthy donors can cure the disease, very few patients get the transplant for a variety of reasons (unlike severe combined immuno deficiency, another HSC disease in which most patients do get transplants). And we know from various sources that editing just 2-5% of alleles can provide benefit to patients (equates to 4-10% of cells, due to the Hardy-Weinberg principle).
But while several groups have tried to make a preclinical gene editing candidate to cure SCD by replacing the disease allele, most efforts have so far met with challenges. Things at first looked very promising in model cell lines, but when people moved to HSCs they found that the efficiency of allele conversion dropped substantially. Efficacious replacement of disease alleles is something of a holy grail for gene editing, but has so far lagged behind our ability to disrupt sequences.
Sequence knockout to cure SCD
In the field of SCD, problems with sequence replacement have prompted efforts to find a way to use sequence disruption to ameliorate the disease. The most promising of these approaches lead to the up-regulation of fetal hemoglobin through a variety of mechanisms, including tissue-specific disruption of Bcl11A (it needs to be tissue-specific because Bcl11A does a lot of things in a lot of cells). Upregulation of fetal hemoglobin has been observed in humans (during Hereditary Persistence of Fetal Hemoglobin, or HPFH), and is protective against the sickling of adult hemoglobin.
Fetal hemoglobin upregulation strategies are very exciting, make for some fascinating basic research, and could eventually lead to new treatments for SCD patients. There are a few people in my lab working along these lines, and several other groups are doing incredible work in this area. But while the editing is easier, there are still some major challenges to clinical translation, mostly due to the new, mostly unexplored biology surrounding tissue-specific disruption of Bcl11A.
Sequence replacement to cure SCD
We decided to go back to the basics, and find out if our work on flap-annealing donors and the Cas9 RNP could bring the “boring” approach of sequence replacement back on the table. In this case, we want to be as boring as possible. I’d rather not get surprised by new biological discoveries about gene regulation while we’re working in patients.
We also wanted our approach to be as general as possible – to develop something that works for SCD but is accessible to clinical researchers everywhere. That would fit the democratization theme of Cas9. Both the nuclease targeting reagent and the approach to allele replacement would be fast, cheap, and easy for everyone to use, so that everyone could ask questions about their own system in 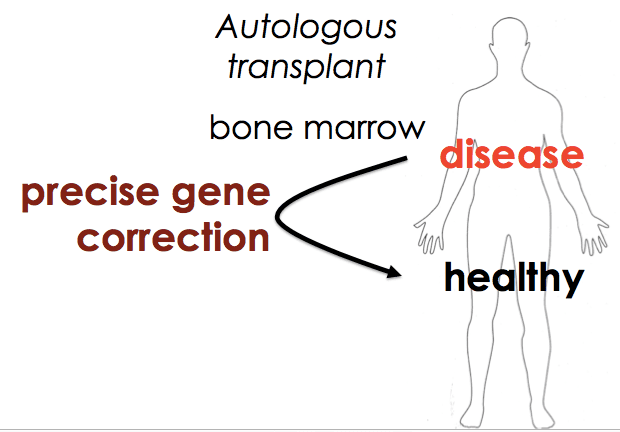 HSCs and maybe even develop gene editing cures for the particular disease in which they’re working.
HSCs and maybe even develop gene editing cures for the particular disease in which they’re working.
This is in contrast to some viral-based editing strategies that seem reasonably effective but are very slow to iterate and have a high barrier to entry. If possible, I’d rather develop something that any hematopoietic researcher or clinical hematologist can pick up and use, with rapid turn around from idea to implementation. That’s the way you get to clinician-driven cures for rare genetic diseases, which is the long-term promise of easily reprogrammable gene editing.
All of the above is why we used Cas9 RNP and coupled it with flap-annealing single stranded DNA donors. We found that we didn’t even need to use chemical protection of the guide RNA or single stranded DNA, though we certainly tried that. In short-term editing we found very high levels of editing, and in long-term mouse experiments we found edits almost five-fold higher than previously reported. I think this is certainly good enough for researchers to start using this method to tackle their own interesting biologies. But for us, there’s still a lot of work to be done before this becomes a cure for SCD.
Next steps towards the clinic
First, we need to try scaling up our editing reagents and method (Cas9, guide RNA, donors, stem cells, and electroporation) to clinical levels, and we also need to source them with clinical purity. This is a non-trivial step, but absolutely critical to eventually starting a clinical trial. Second, we need to establish the safety of our approach. We found one major off-target site while doing the editing, and it lies in a gene desert. But just because it’s not near anything that looks important doesn’t mean we don’t need to do functional safety studies! (see my previous blog post about safety for gene editing for more on this) We have a battery of safety studies planned in non-human models, and we want to take a close look to see whether we can take the next step into the clinic.
My collaborators and I are committed to the clinical application of gene editing to cure sickle cell disease, and we hope to start a clinical trial within the next five years. But the data will guide us – we want to be very careful, so that that we know that the cure is not worse than the disease. There are many moving parts involved in these translational steps and some of them are a little slow, so please stay tuned as we try to bring a breakthrough cure to patients with SCD.



